由于本科时,在任老师的指导下,了解并学习了关于理论计算在单分子配合物分子中的应用,因此小入了一点门。到了研究生阶段,也就想着尝试将理论计算结合到自己的工作中,我的研究课题是基于钌配合物的探针和光敏剂设计,因此主要做的工作是基于金属配合物发光的理论研究。但是,我们课题组有不少的同学的研究方向为基于过渡和稀土金属的分子笼或MOFs的催化性能研究和识别性能研究,有时也需要相应的理论计算支持。而关于这个方向的计算与我自己的研究方向是有所不同的,因此想着写一篇简单的博文,整理一些自己认为重要的点方便自己回顾,同时期望帮助组内的同学可以快速的上手操作并获取自己想要的数据。
前期准备
设备:服务器(不建议使用个人电脑进行提交任务)、个人电脑(用于前期的输入文件准备和数据可视化与处理)
服务器系统:Linux系统(Windows的Gaussian的运行效率没有Linux的好)(听说的,具体那看到的忘了,反正建议使用Linux系统)
个人电脑系统:什么系统都行(Windows、Mac或Linux),只要自己会用(反正我用的Windows)
软件:Gaussian(用于执行计算任务,安装于服务器),GaussView(用于数据的可视化与处理,安装在于个人电脑),Multiwfn(用于数据的处理,安装于个人电脑和服务器都行,只要再相应的系统会用就行)
个人技能:应该知道如果操作Linux系统(非桌面系统),以及如何建模、准备Gaussian输入文件和提交Gaussian任务(后面我还是会写一点的)
Linux系统的操作
使用Linux应该了解的操作应该有如何进入文件夹、如何退出文件夹、如何显示当前文件夹的文件、如何copy文件、如何删除文件和如何删除文件夹等等,反正就是在Win系统中常见的操作应该学会如何使用命令行的方式进行操作。
文件夹操作
为什么要进行文件的操作捏,因为在Linux系统中,进行相关的任务操作都是在当前的文件中进行,并不能在文件夹1中去操作文件夹2中的任务,为了操作文件夹2中的任务,需要通过文件夹的切换,到文件夹2中后才能进行任务操作。因此有必要了解下面写的一些简单的命令,当然Linux系统的命令是丰富的,如果需要完成一些下面没有提到的操作,可以直接在浏览器搜索Linux命令大全(应该会出现很多,捡一下觉得顺眼的学习一下就好了)。
创建文件夹
1 | mkdir 文件夹 |
就像上面写的那样,很简单的。
进入文件夹
1 | cd 文件夹 |
只需要cd文件夹就可以进入下一级文件夹。
返回上级文件夹
1 | cd ../ |
在cd的时候使用两个点和一个反斜杠就可以返回到上一级文件夹。
当然我们也是可以使用cd进入到多层文件夹里面,只需要如下操作就可以。
1 | cd 文件夹1/文件夹2 |
往上返回多层文件夹同理,操作如下。
1 | cd ../../ |
删除文件夹
1 | rm -rf 文件夹 |
加上-rf是因为有时候文件夹内可能还有文件(非空文件夹),直接使用rm系统是不让删除的,因此加上-rf可以解决这个问题。
根据上面的操作,基本可以实现在服务器中的文件夹的操作。
文件的操作
由于本博文主要服务于理论计算,因此文件的操作主要针对文本文件。(一般在Linux系统中,文本文件使用Vim操作)
文件的新建
1 | vi 文件名 |
文件名一定要带后缀!!!
如txt文件的形式一定是“文件名.txt”,Gaussian的文件形式为“文件名.gjf”。
使用上面的命令后,就可新建一下文本文件,并进入这个文本文件,这个时候文本文件处于阅览模式,如果想编辑这个文本文件,则需要再英文输入模式下敲击键盘的i,这个时候可以再最下端看到编辑模式为“inset”,这个时候就可以进行文本信息的编辑。当完成文本的编辑后,请敲击ESC退出编辑模式,再输入:wq,就是保存该文本并推出文本文件。
文件的复制
1 | cp 文件名 目标文件夹 |
也很简单。
文件的删除
1 | rm -f 文件名 |
-f的加入是为了强制删除该文件。
根据上面的操作,基本可以满足当前的工作需求。
Gaussian软件的操作
Gaussian是提供量子化学计算的主要软件,本博客所涉及的所有量化计算都是使用该软件完成。因此,这部分主要讲的是Linux中Gaussian的操作,即任务的提交,查看和删除。
任务提交
1 | g16 文件名.gjf 文件名.out |
g16是指使用的Gaussian软件版本为Gaussian16,如果服务器安装的为Gaussian09则这个地方需要该为g09。文件名.gjf为Gaussian的输入文件,文件名.out为Gaussian的输出文件。(对于输出文件,也可以将out后缀改为log后缀)
如果想使当前任务为后台运行(后台运行的好处是可以不用停在软件执行页面,操作者可以进行别的操作,即使退出账号后也是可以继续执行后台任务的,如果是非后台运行任务,退出服务器时,任务也就一起停止了),只需要在其命令后面添加“&”即可,操作如下。
1 | g16 文件名.gjf 文件名.out & |
如果服务器使用的为“Slurm”作业调度系统”进行任务的管理,则需要将上述任务提交命令填入相应的脚本文件中,通过提交脚本来实现脚本文件。
任务查看
1 | top |
top命令经常用来监控Linux的系统状况,因此也可以用于监视提交的Gaussian任务,对于成功提交的任务都会产生一个对应的PID号,该PID号对应的USER就是提交任务的账号名称(可以用于核对该任务是否为自己提交的任务),同时对应COMMAND显示的就为Gaussian调用的任务程序(其实可以通过这个任务程序判断Gaussian进到到那个模块)。
当然ps也是可以查看的任务的,但是提供的信息没有top提供的全,同时,在退出一次账号后,进入服务器后使用ps是不能查看到后台执行的任务的。
如果服务器使用的为“Slurm 作业调度系统”进行任务的管理,只需要输入squeue即可查看,它会提供一个唯一的JOBID,同时提供USER(提交用户)和ST(任务状态)信息。
任务删除
1 | kill PID |
如果想停止正在执行的Gaussian任务,这时就可以使用kill来杀死这个任务,PID可以通过top来查询。
如果服务器使用的为“Slurm 作业调度系统”进行任务的管理,则需要使用scancel命令,操作如下。
1 | scancel JOBID |
JOBID可以使用上节提到的squeue来查询。
GaussView软件的操作
GaussView是一款由Gaussian公司出版的搭配Gaussian使用的可视化软件,使用该软件可以帮助软件使用者快速的建模并建立输入文件,同时可以用于Gaussian输出文件的分析。
下面将简单的介绍软件的界面和操作,更详细的信息介绍可以查看官方文档,以及直接去Bilibili搜索相关教程,讲的都比此处详细和便于理解(我认为还是视频比较形象化)。
软件界面的介绍:
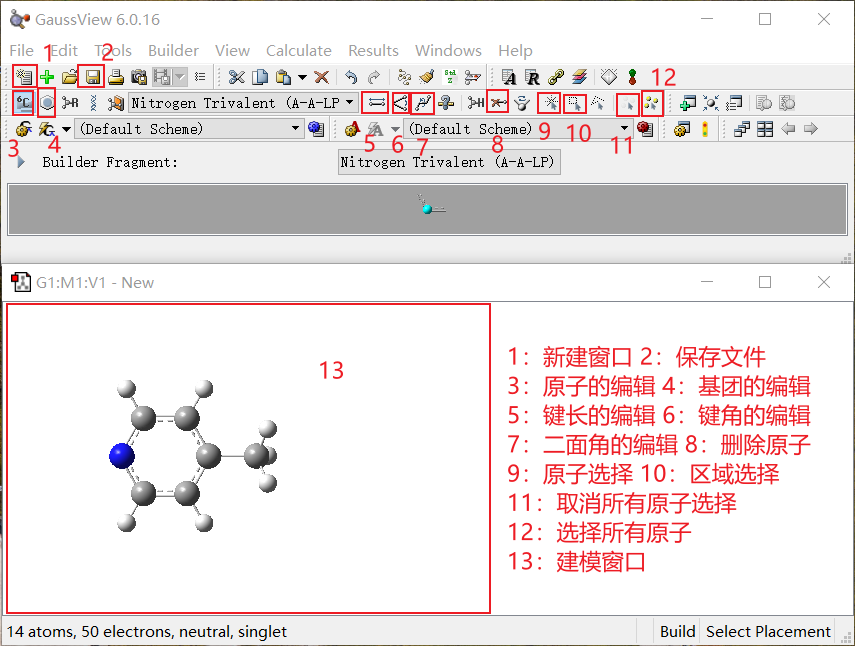
通过使用以上的功能选项,基本可以搭建出大部分的分子结构。
Gaussian输入文件的构建
对于Gaussian的计算,Gaussian的输入文件是必不可少的,其主要的目的告诉软件执行什么样的操作。Gaussian输入文件一般通过两种方式构建,一是通过GaussView软件建模并设置相关的计算信息,二是通过编辑文本文件的方式编辑输入文件(只要有分子结构的原子坐标,这个方法是更方便的)。一般情况下,建议上面两种方法结合使用,通过GaussView进行分子结构的建模,再通过文本编辑输入自己想让Gaussian执行的任务命令。
由于网上有大量类似的内容,此处就不再详细整理。
此处推荐一篇博文供学习,来自Jiang Shen的Gaussian 笔记。
由于我的课题组主要做的分子结构都含有金属原子,因此在此整理一些含金属原子的体系应该如何编辑输入文件。
以含钌原子的配合物为例
1 | %chk=Rucomplex.chk ## 申明生成chk文件 |
注意点:
- 关于泛函和基组的选择,如果自己也不知道对于自己的体系使用什么样的泛函和基组,最好的方法就是参考文献中类似体系使用的泛函和基组,照抄到自己的计算体系中。
- 在计算内容中,由于计算体系含有金属原子,需要使用
genecp关键词,申明该体系对金属原子使用膺势基组(Los Alamos赝势或称Lanl赝势、Stuttgart/Dresden赝势或称SDD赝势、SBKJC赝势也称CEP赝势等等),同时对原子基组的定义由原先的计算内容行,变为在分子的直角坐标后。 - 对于含金属原子的体系,一般在移除抗衡阴离子后,通常不是中性体系的,因此这个时候的电荷需要进行相应的计算,不能像中性分子体系那样使用0电荷一概而论,同时含有金属原子的体系,基态下的自旋多重度也不在一定为1,这个需要进行相应的计算来确认。
- 关于原子坐标的获取,对于有晶体结构的体系,可以直接使用晶体结构作为计算的初始结构(这个结构是需要优化的),也可以使用建模软件进行搭建,例如GaussView。
- 对于稀土元素,建议使用SDD基组中的大核膺势,一般对于过渡金属使用的SDD其实代表的是小核膺势。
常见任务的关键词
这部分由于个人水平有限,只能粗浅的写写,期望起到抛砖引玉的效果。关于一些更具体的操作和技巧,建议参考Gaussian的官方文档或者Sob老师的相关博文。
结构优化
对于所有的结构,都应该首先被优化,以获得一个最稳的结构点,这样才能保证后面的计算的有效性。
一般计算需要使用的计算内容为:
1 | # b3lyp 6-31g(d) opt |
上面的第一项为泛函(有的地方也称为方法),第二项为基组,第三项为结构优化的关键词。
对于优化完的结构,如果想检验是否为能量最小值点,只需要基于原来结构优化后的结构再提交一个频率计算的任务,即可以通过计算结果判断。
一般计算使用的计算内容为:
1 | # b3lyp 6-31g(d) freq |
对于频率计算必须保持与结构优化的泛函和基组一致。
当然,如果想同时提交上面两个任务也是可以的,操作如下:
1 | # b3lyp 6-31g(d) opt freq |
红外光谱
这里提到的红外光谱的计算其实和上面提到的频率计算是一样的,当优化完结构后,进行如下操作即可:
1 | # b3lyp 6-31g(d) freq |
紫外光谱
进行紫外光谱的计算,其实是对分子激发态的计算,因此,原来的密度泛函理论(DFT)计算变为了含时密度泛函理论(TD-DFT)计算。
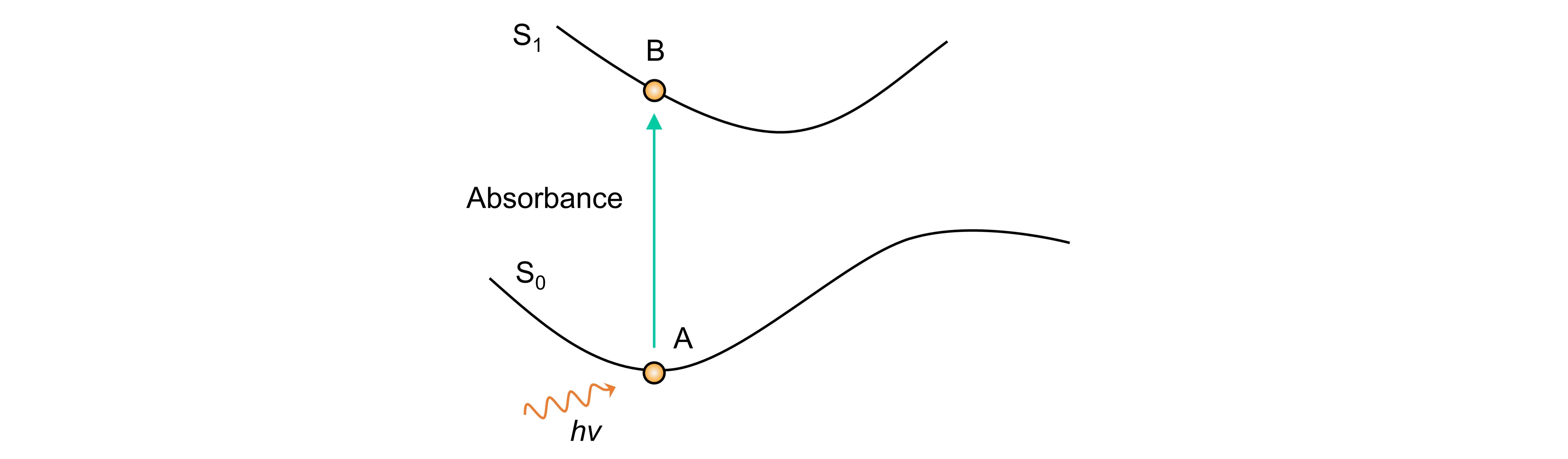
如上图所示,基于S0态的最优结构,不断进行电子激发,使电子跃迁到更高的单重激发态,这也是紫外吸收计算的原理。
这部分操作也非常简单,基于原来结构优化后的结构,提交一个紫外吸收计算的任务即可。
一般计算内容为:
1 | # b3lyp 6-31g(d) td=(singlet, root=1, nstates=3) |
td代表进行TD-DFT计算,其中singlet代表进行激发态为单重态的计算(常说的紫外吸收,其实就是单重态激发),root=1代表感兴趣的态为S1,nstates=3代表共计算能量最低的3个激发态的信息(如激发能、跃迁电/磁偶极矩、振子强度、组态系数等)。其中,singlet为默认关键词(可以不写),root的默认值为1(如果想设置感兴趣的态为2,则需要写root=2),nstates的默认值为3(一般建议设置值比研究的激发态多3个态比较合适)。
荧光光谱
荧光光谱的计算其实也可以一种紫外吸收计算,只是这里计算使用的结构不在是S0的最优结构,而是S1的最优结构。
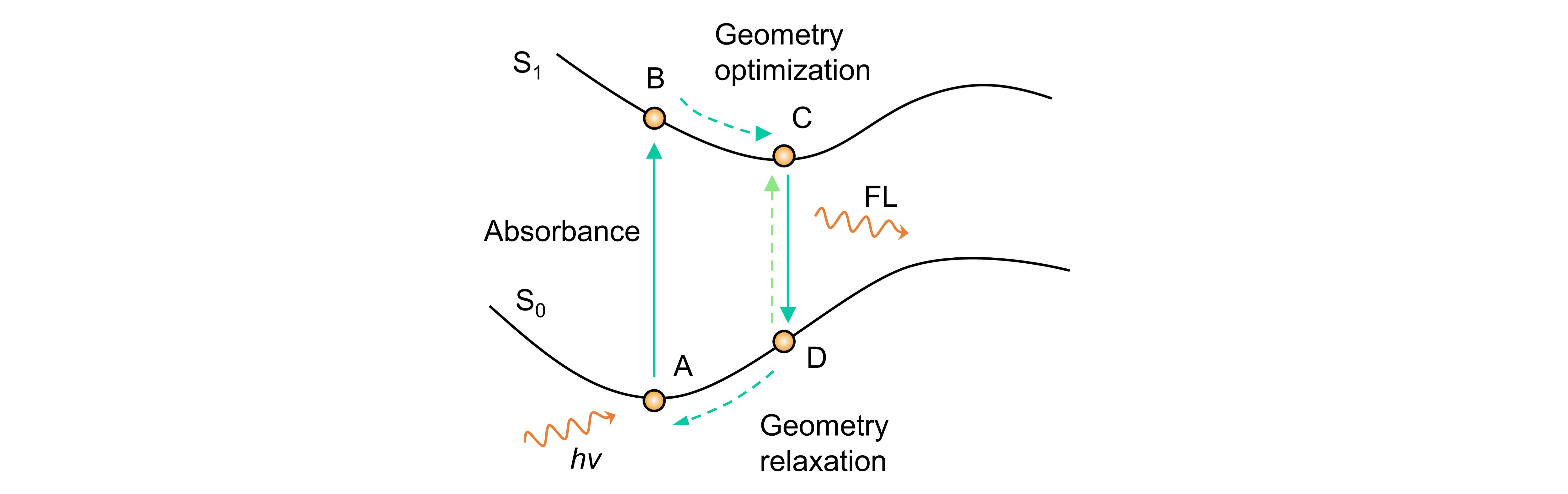
如上图所示,荧光发射需要激发的电子经过一系列的内转换,最后到达S1态能量的最小值点(C点),再通过辐射过程的方式返回S0态(D点)。因此,再计算中有必要寻找S1态的最小值点,然后基于这个最小值点的结构进行td计算。
首先需要获得S1的能量最小值点的结构,操作如下:
1 | # b3lyp 6-31g(d) td opt freq |
S1态优化的初始结构为S0态优化的最终结构。
在获得S1态的能量最小值结构后,基于上一步获得的S1态结构就可以进行激发态计算,操作如下:
1 | # b3lyp 6-31g(d) td |
这个时候,计算产生的S1态对应的就是荧光发射的信息。
上面列举的是一些常见的计算任务,对于一些其他的计算任务,在量子化学公社应该都能查到相应的结果,以后有空我也将会进一步进行补充。
书籍阅读建议
Gaussian官方出版的《Exploring Chemistry with Electronic Structure Methods》第二版
Gaussian软件的用户手册
《关于入门高斯的一个小小指南 V1.6》by Paramecium86
最后
写了这么多,确实也累了,也真的没想到,我尽然还能写出一篇这么长的博文,实属不易。(虽然,可能里面大多数为流水账,望见谅)