UCSF ChimeraX(或简称ChimeraX)是继UCSF Chimer之后,生物计算、可视化和信息学资源(RBVI)的下一代分子可视化程序(这句话是从官网上抄的)。ChimeraX作为一款功能强大的生化分析和可视化软件,对于我这样一个学化学的,是不太会那些高端操作的,也没那能力,但最近觉得ChimeraX做的图质感很好,随产生了用该软件画反应机理计算中的分子结构图的想法。但我是小白呀,从没接触过该软件,对于分子的调整完全不了解。本着边学边做笔记的想法,随产生了写这篇博文的想法。
软件的安装
软件的下载,来自官网 UCSF ChimeraX Home Page。
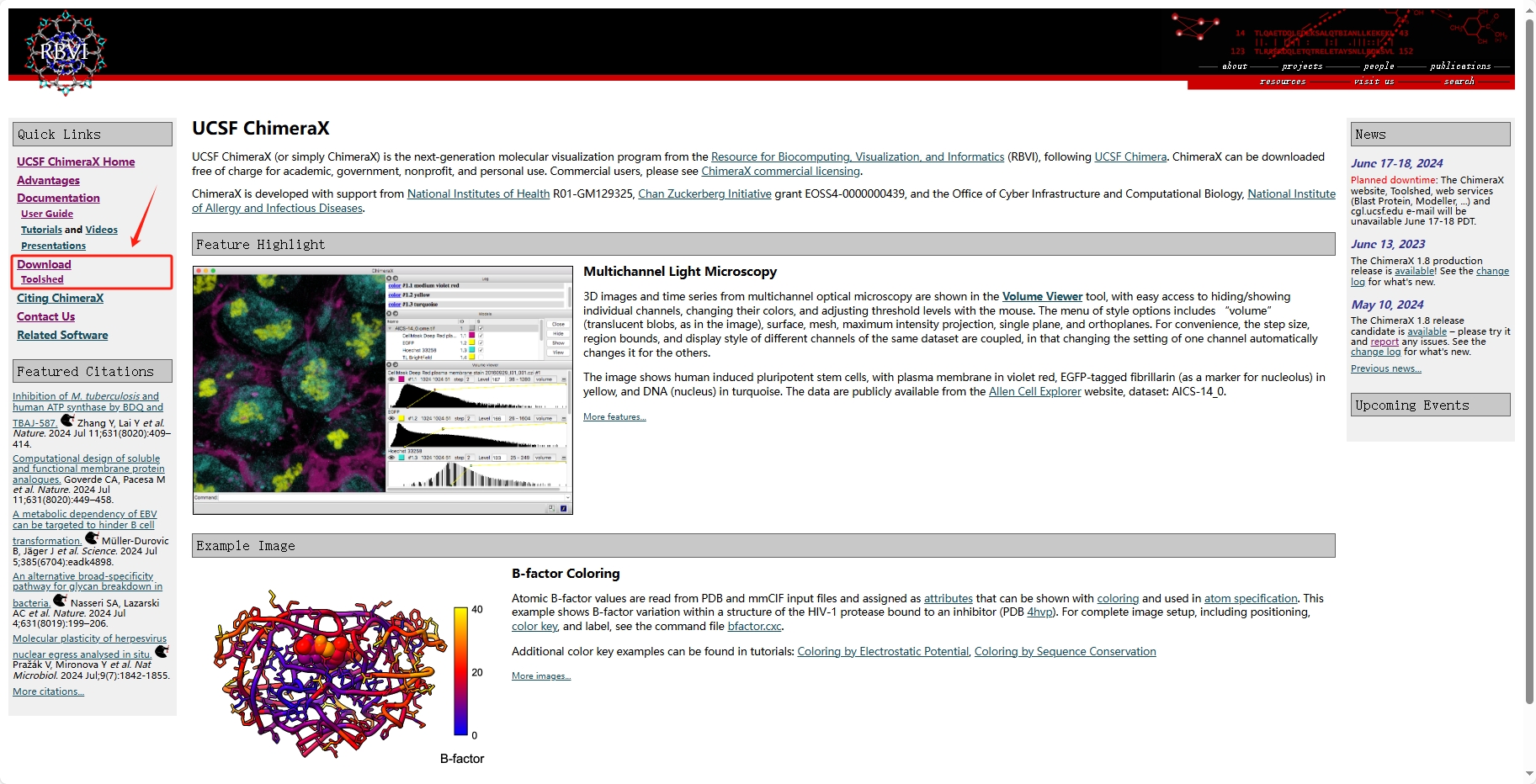
点击Download,进入软件下载页面 Download UCSF ChimeraX。
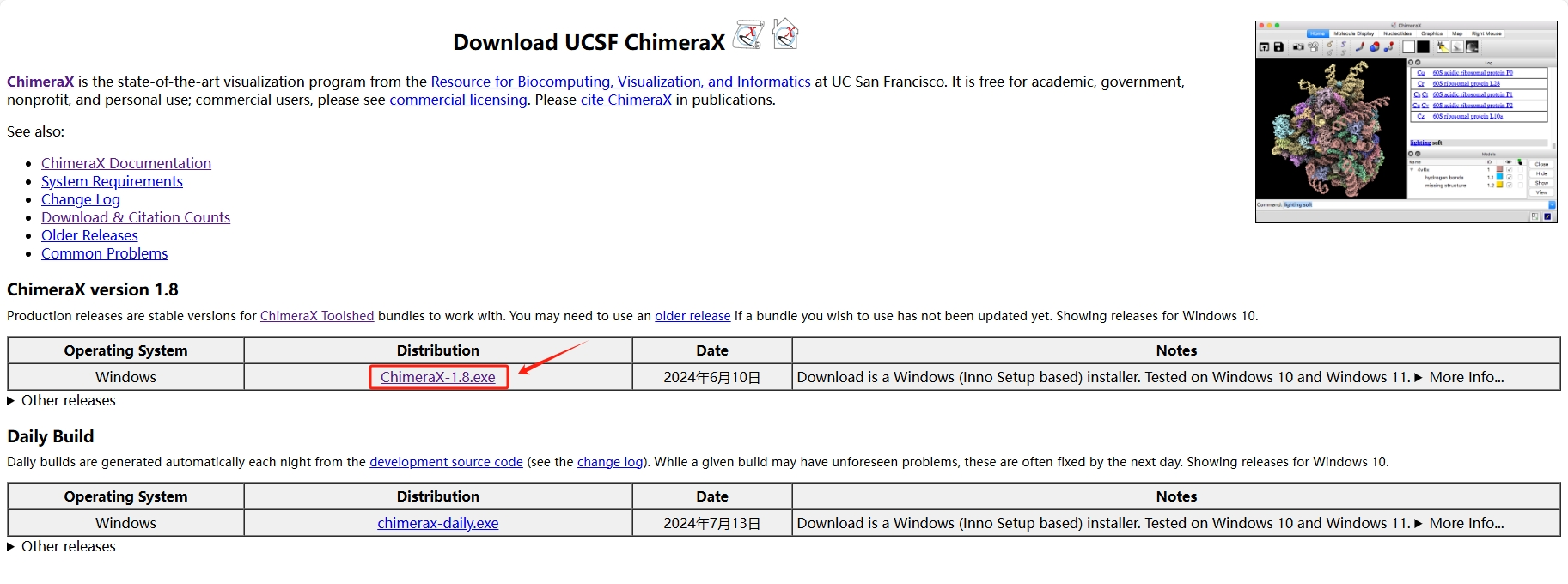
此处下载的版本为Windows ChimearX-1.8.exe,点击上图红色框的位置,进入到UCSF ChimeraX的非商业软件许可协议,点击最下方的 Accept,即可下载该软件。
根据作者要求使用该软件发表文章应添加acknowledgment和cite。
include an acknowledgment similar to the following:
Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
cite one or more of the ChimeraX references, mainly:
UCSF ChimeraX: Tools for structure building and analysis. Meng EC, Goddard TD, Pettersen EF, Couch GS, Pearson ZJ, Morris JH, Ferrin TE. Protein Sci. 2023 Nov;32(11):e4792.UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. Protein Sci. 2021 Jan;30(1):70-82.
UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE. Protein Sci. 2018 Jan;27(1):14-25.
Publications include papers, posters, films, videos, exhibits, and artwork. The home page https://www.rbvi.ucsf.edu/chimerax can also be cited.
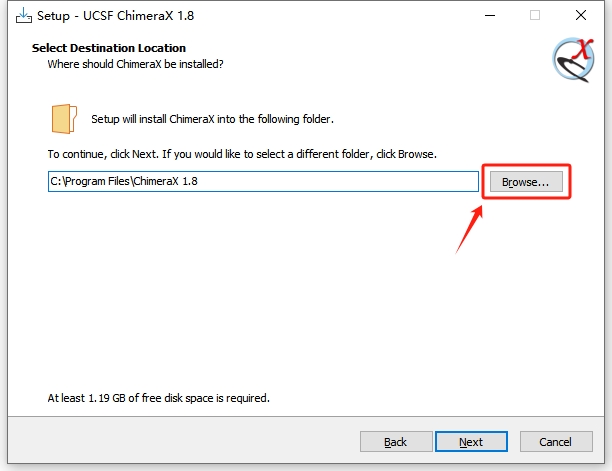
下载完软件安装包后,对于Windows系统直接双击即可,在安装的过程中,如果有修改安装路径的需求,应自定义安装路径(点击下图红色框选择安装位置)。
正常安装完后,双击桌面图标,即可使用该软件。
软件的使用
文件的准备
最近刚好做了一个醛胺氨缩合的过渡态,就以这个有机反应进行举例,反应方程式如下:

对于这个反应的进行过程,从量子化学的角度由应该如何考虑呐?
那么就需要思考这个反应的醛基是如何进攻苯胺,如果找到这个过渡态,也就找到了常说的反应能垒。
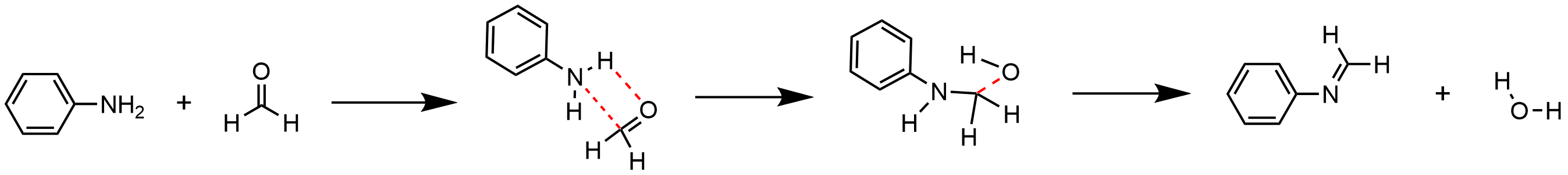
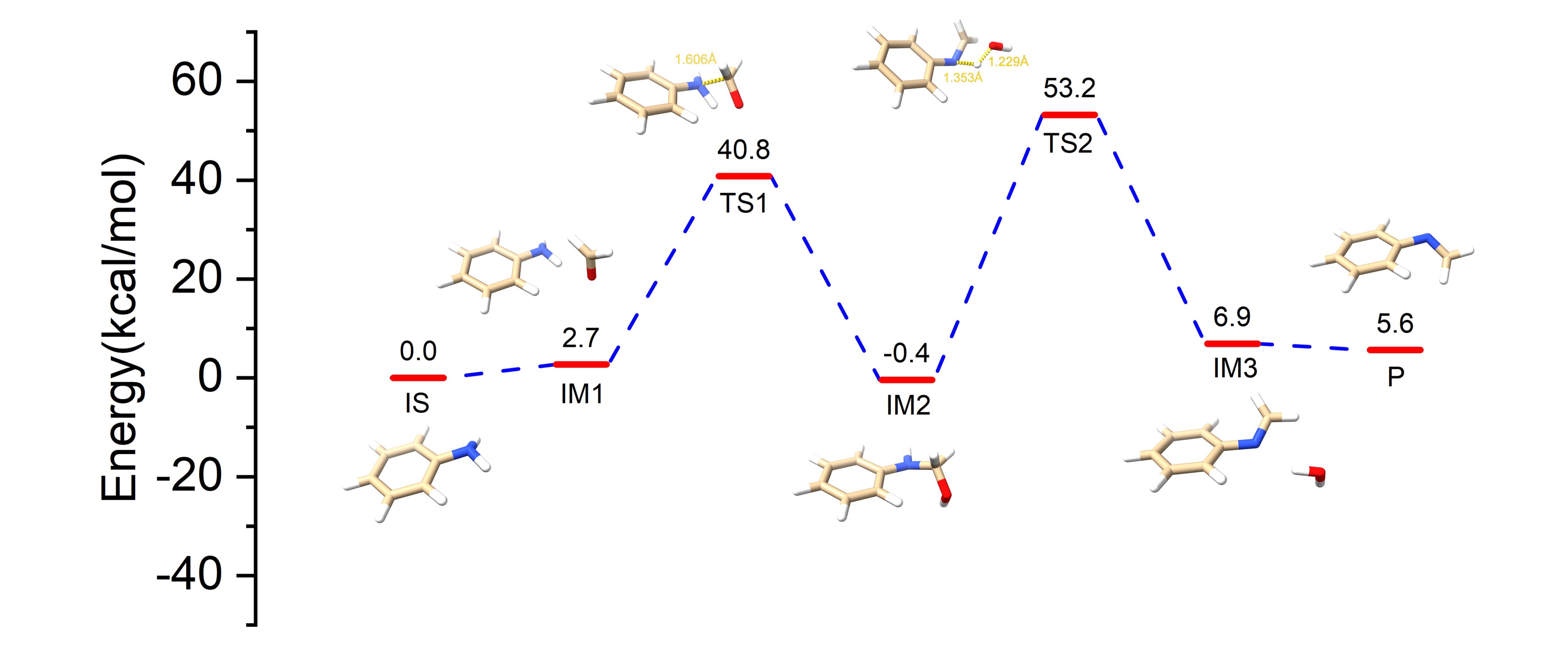
如上氨基氮进攻羰基碳,形成中间态,再通过质子交换,将羰基碳氢转移到羰基氧上,最后脱水形成亚胺。
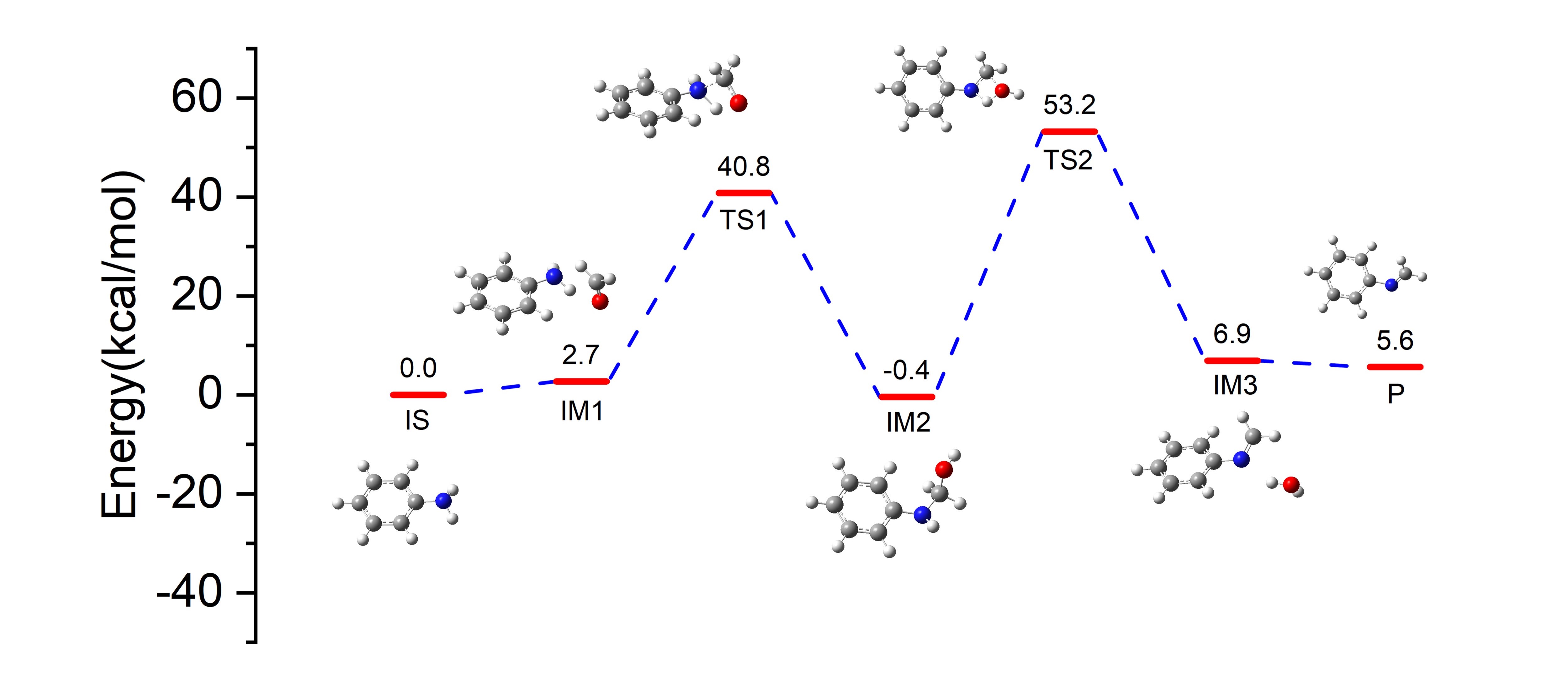
上图是通过Gaussian找到的两个过渡态,频率计算验证只有一个虚频,最后通过IRC找到了对应的始态和终态,与推测的结果一致,证明了这些过度态的合理性。
在以上一通操作下,获得了一个始态、一个终态、两个过渡态和三个中间态,虽然也成功画出了反应路径的能级图,但总觉得GaussView导出的分子结构图不是很美观(肯定也能用拉),因此开始本博文的内容,进一步的美化。
文件格式的转换
一般Gaussian计算过的输出文件为out文件,需要将out文件转化为pdb文件。
此处使用卢天老师的Multiwfn软件进行格式转换。
文件的导入
双击桌面的ChimeraX软件图标,进入到软件界面。
将刚才导出的pdb文件拖入软件的灰色窗口,这里TS1的pdb文件为例。
操作:
- 按住鼠标左键移动鼠标,可以旋转分子模型;
- 按住鼠标滚轮移动鼠标,可以平移分子模型;
- 旋转鼠标滚轮,可以放大或缩小分子模型。
对于这个分子,现在我对它的模型是满意的,但是它在显示上是有一个错误的地方,即氨基氮与醛基碳之间的键不应该是实键,因为是过度态,应该为虚键这种伪键才对。以下内容将针对这个目的进行操作实现。
分子模型的修改
根据在计算化学公社查到的一篇名为《Chimera如何自定义两个原子之间的连接方式呢》的博文,获得了一种修改两个原子之间连接方式的方法。
博文内容为:
首先第一步,Alt+鼠标左键选择想要改变的键,点击右下角的放大镜图标,打开Inspect Selection窗口;第二步选择Molecule model选项,将下面的line style改成自己想要的模式;第三步选择Bond选项,将bond style改为wire,将halfbond mode关闭后可以在color处更改键的颜色;第四步退出选中模式即可,图中的实键已经变为了自己想要的虚线。另外这个过程是可逆的,也有其他的选项可供大家设置,谢谢大家。
我也尝试了改博主写的的修改方法,但是在做到第二步的时候,就遇到了问题,在软件的Inspect Selection窗口没有Molecule model选项,因此没能推进下去。(我电脑安装的软件也没有显示第一步所说的放大镜图标,该窗口的打开,我是通过home选项下的Selection部分的inspect打开的。)
由于上面的方法没有行通,因此又继续寻找了新的方法。
其实上面也讲了,对于该分子只是希望在两个原子之家形成以虚线形式显示的伪键。因此,首先去查询了如何创建一个新的伪键,官方文档给出的是通过命令行的方法,但是经过了多次尝试,并没有成功执行目标命令,这条路随止。(后期如果我看懂了官方文档,我在补充这部分内容)
无意间我看到了distance这个选项,想到我本来的目的就是创建一个虚线的伪键,以及显示对应的长度,那么是不是可以通过distance来实现我的目的,随进行了尝试。
第一步,按住ctrl键使用鼠标选中需要修改的键(选中后会出现绿色边框);
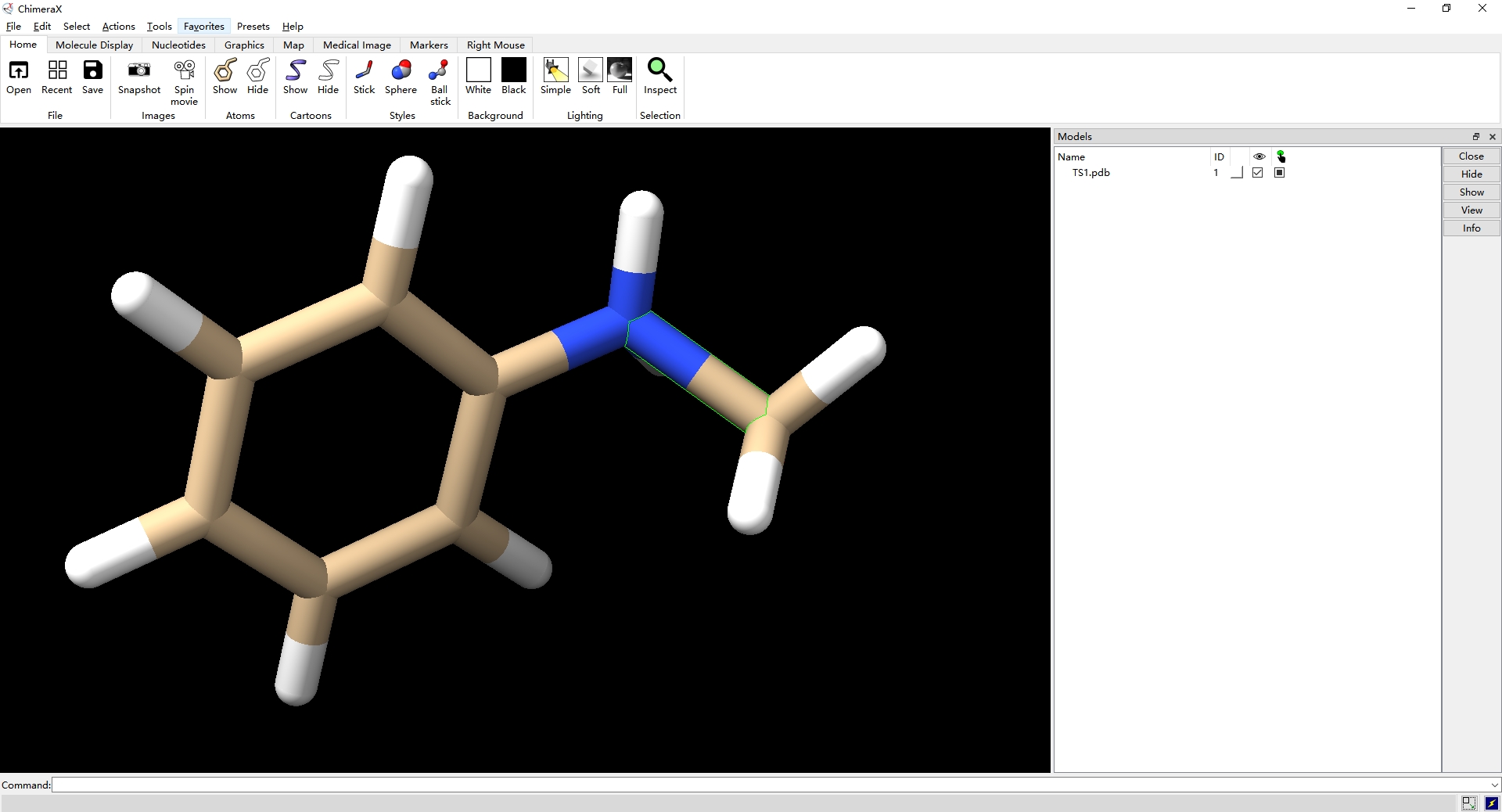
第二步,在点击Actions下的Atoms/Bonds选项的Hide,隐藏该碳氮键;
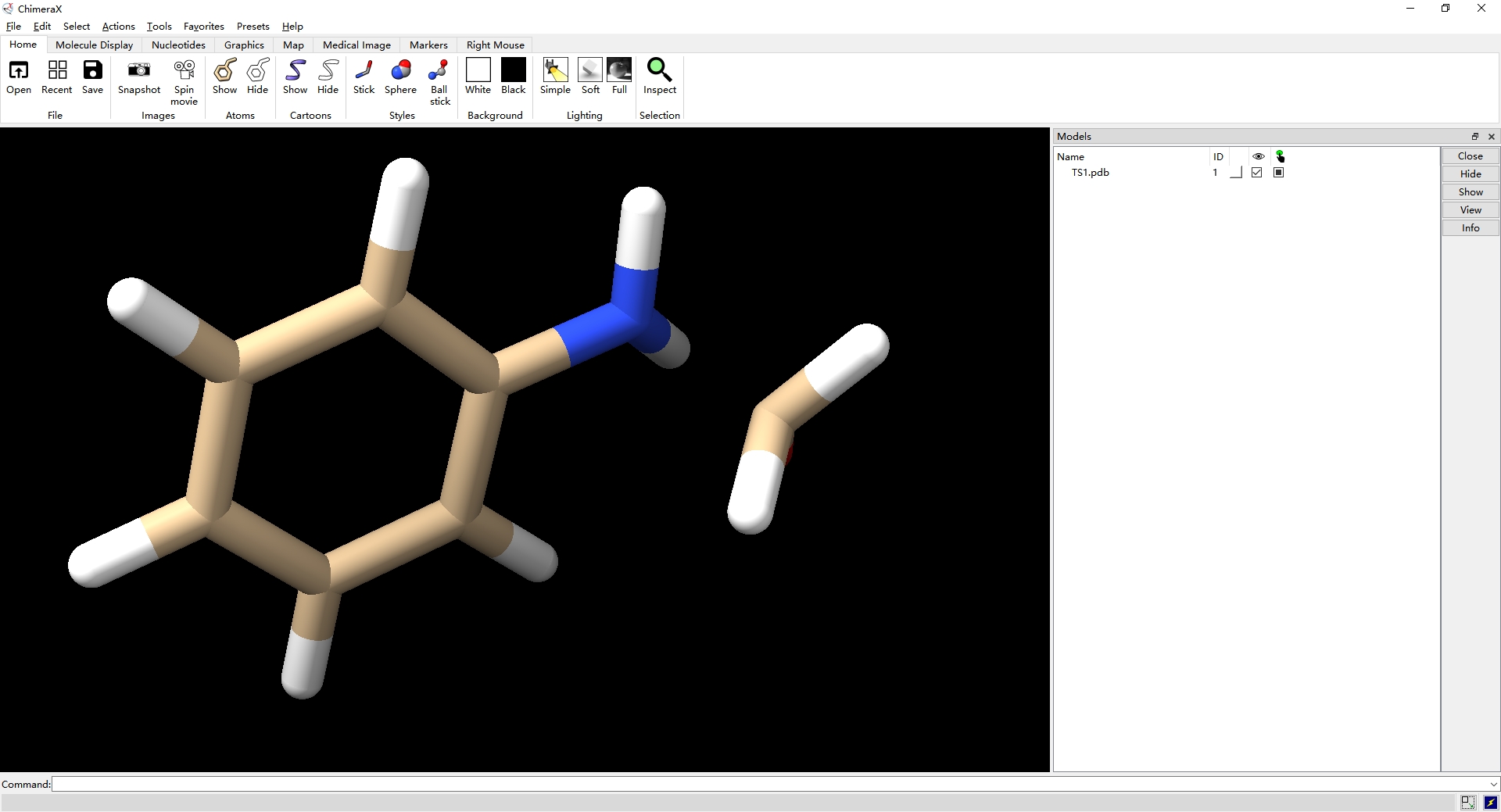
第三步,点击Right Mouse选项下的Annotation部分的Distance(选中后该icon会出现绿色背景),接着同时按着Ctrl和Shift,选中氨基氮原子和醛基碳原子,再右击鼠标,就会产生虚线的伪键和键长参数;
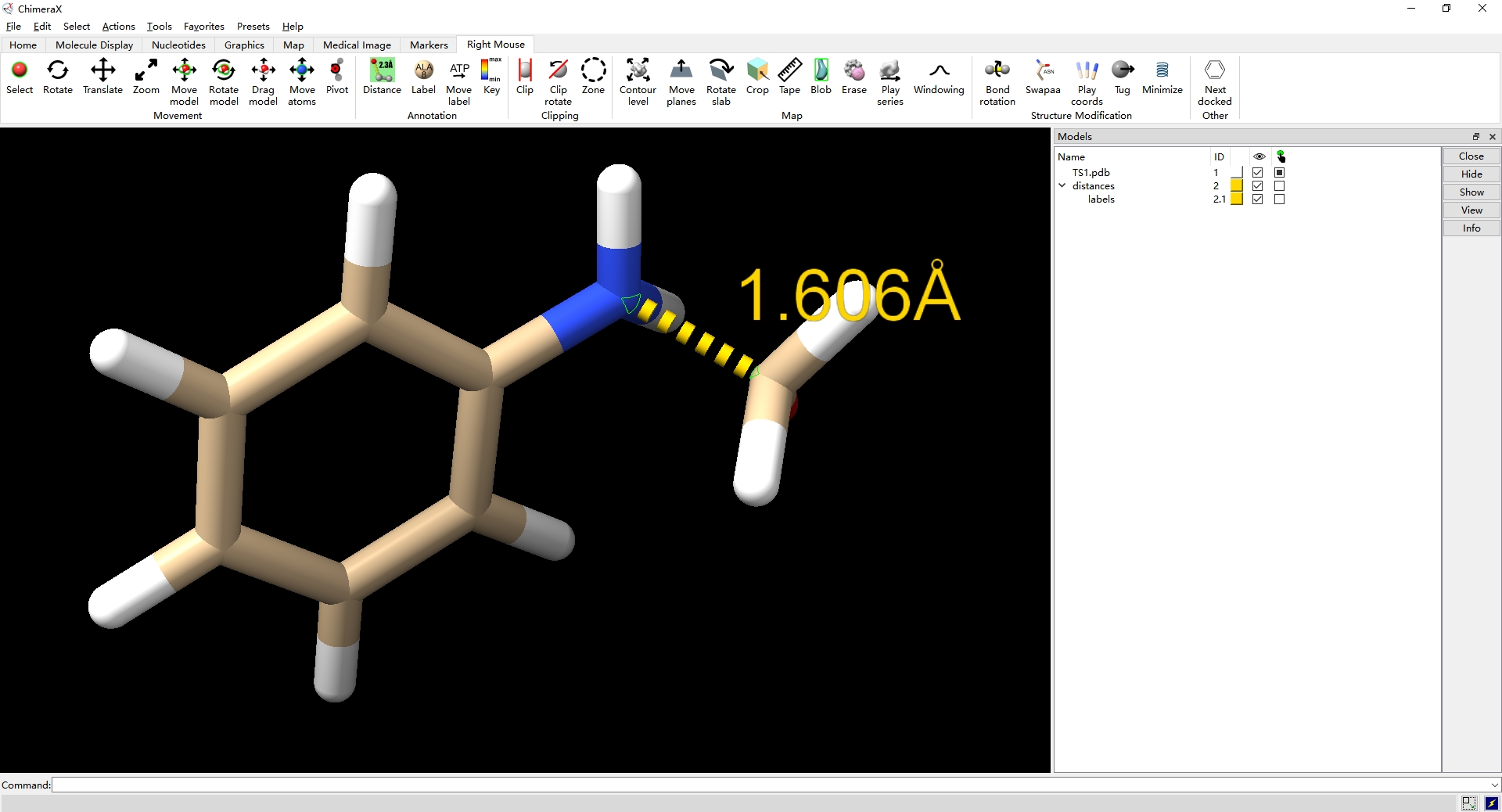
第四步,按住Ctrl后,选中虚线伪键,点击home选项下的Selection部分的inspect打开Selection Insqector窗口;
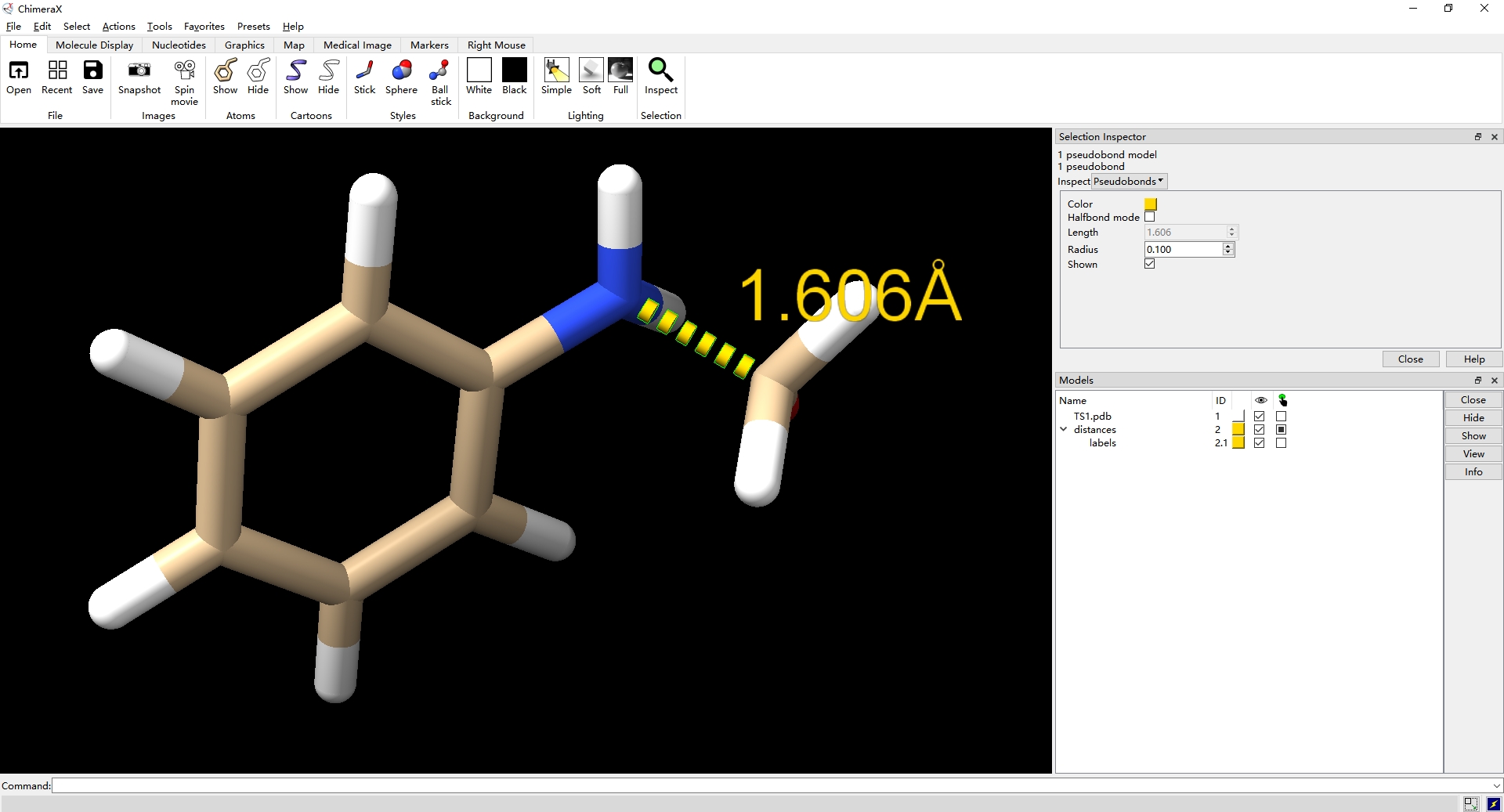
第五步,在Selection Insqector窗口将Inspect的Pseudibonds改为Pseudibond models,通过修改Color可以改变键的颜色,修改Dashes可以改变伪键的分段数,修改Radius可以改变伪键的半径。(这里我只改变了Dashes,因为感觉原先的分段太密集了。)
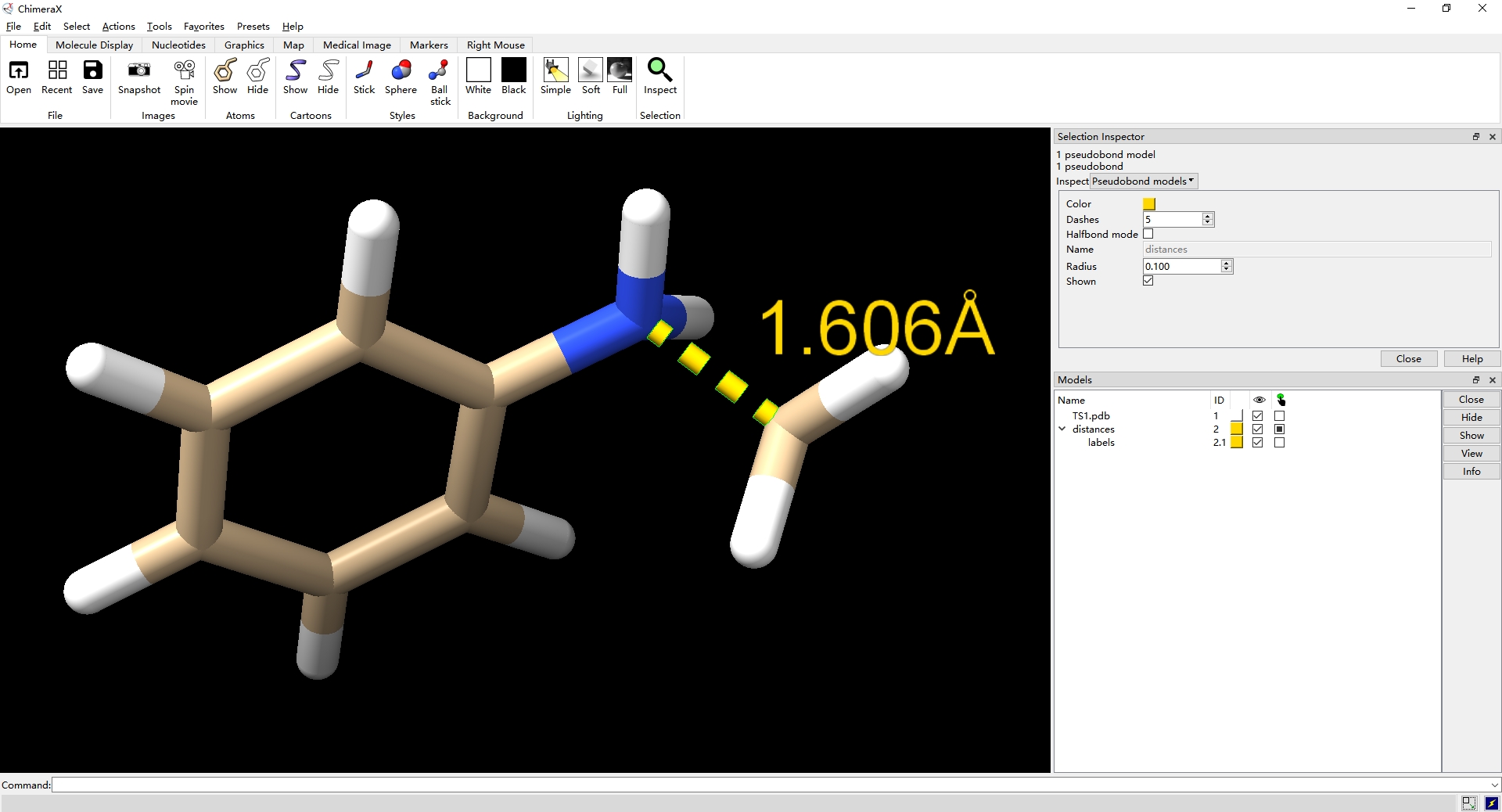
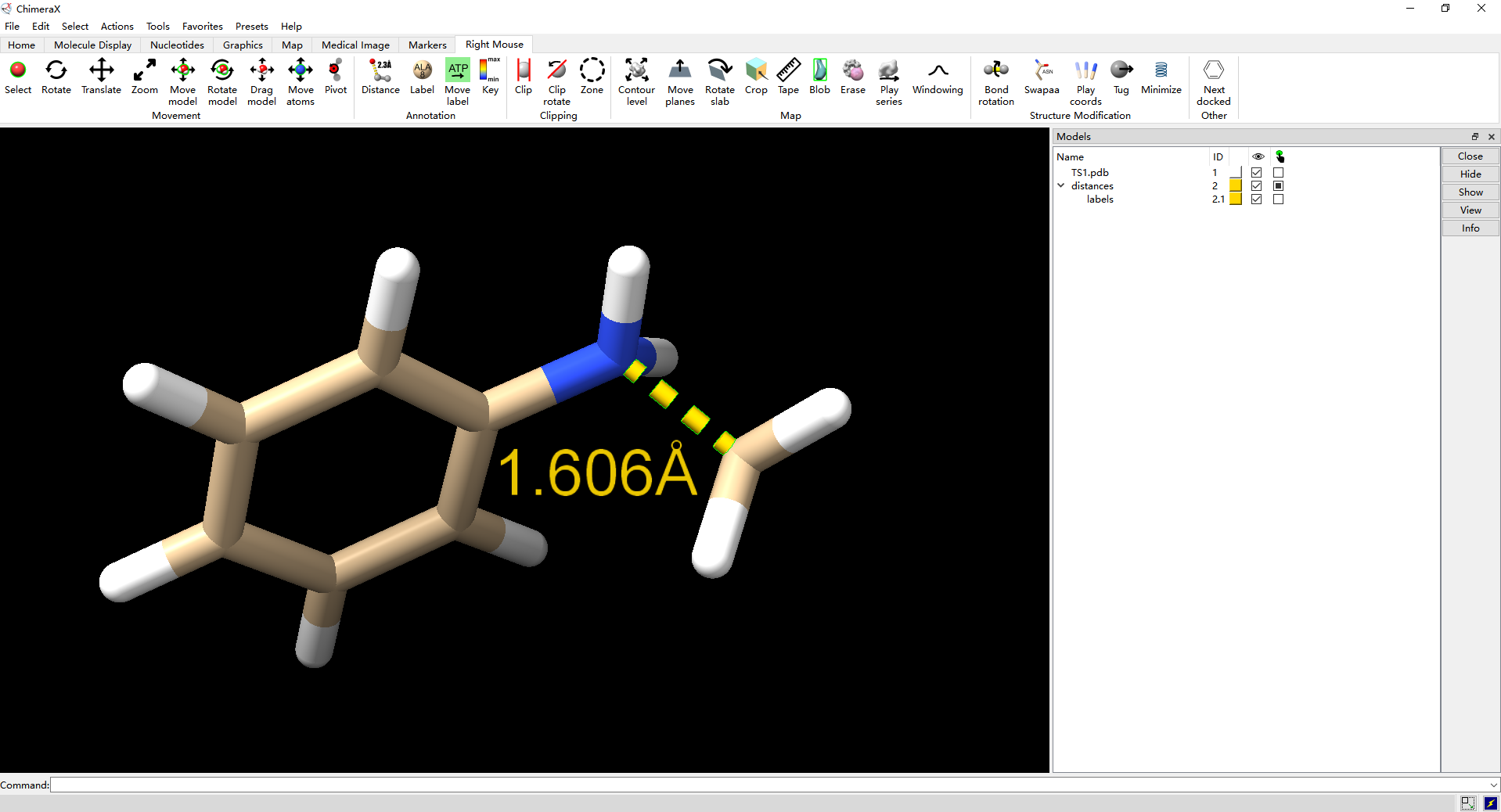
第六步,点击Right Mouse选项下的Annotation部分的Move label(选中后该icon会出现绿色背景),然后将鼠标移动至label处并按住鼠标右键,即可移动label位置;
另一种方法,在最下方的Command命令窗口输入label offset 0.1,0.2,0,来修改标签的位置。
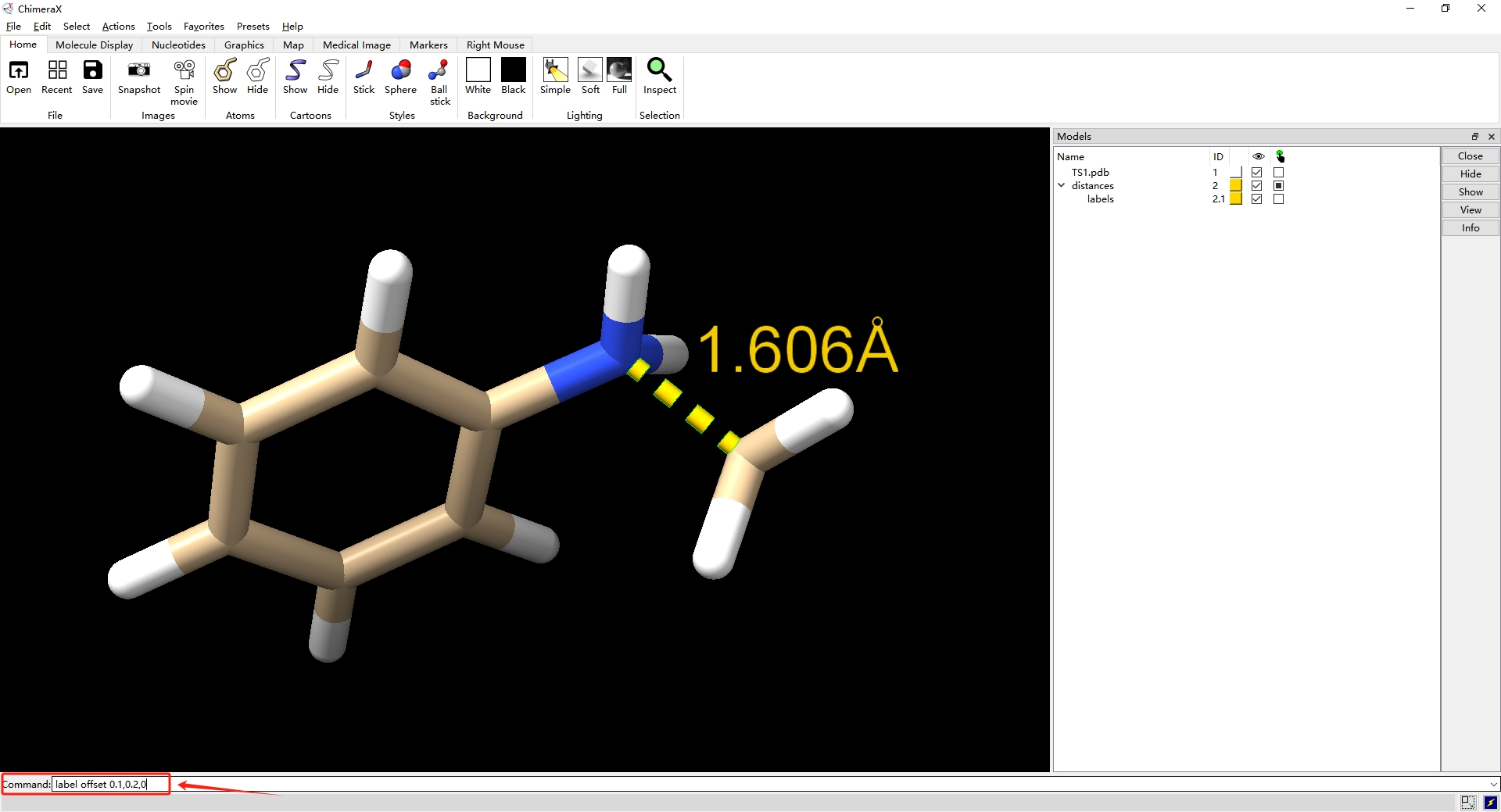
offset将标签的绝对偏移量指定为屏幕坐标系中沿x、y和z轴的三个逗号分隔值x、y、z,以数据的距离单位(通常为Å)表示。偏移量是标签矩形左下角相对于标记项目中心的位置。可以给出负值。默认偏移量为:0,0,0。
注:第二种方法,通过上述命令只适合单label,对于多label的位置调整,还请自行查阅官方文档。
第七步,点击home选项下的Background部分White,修改背景为白色;
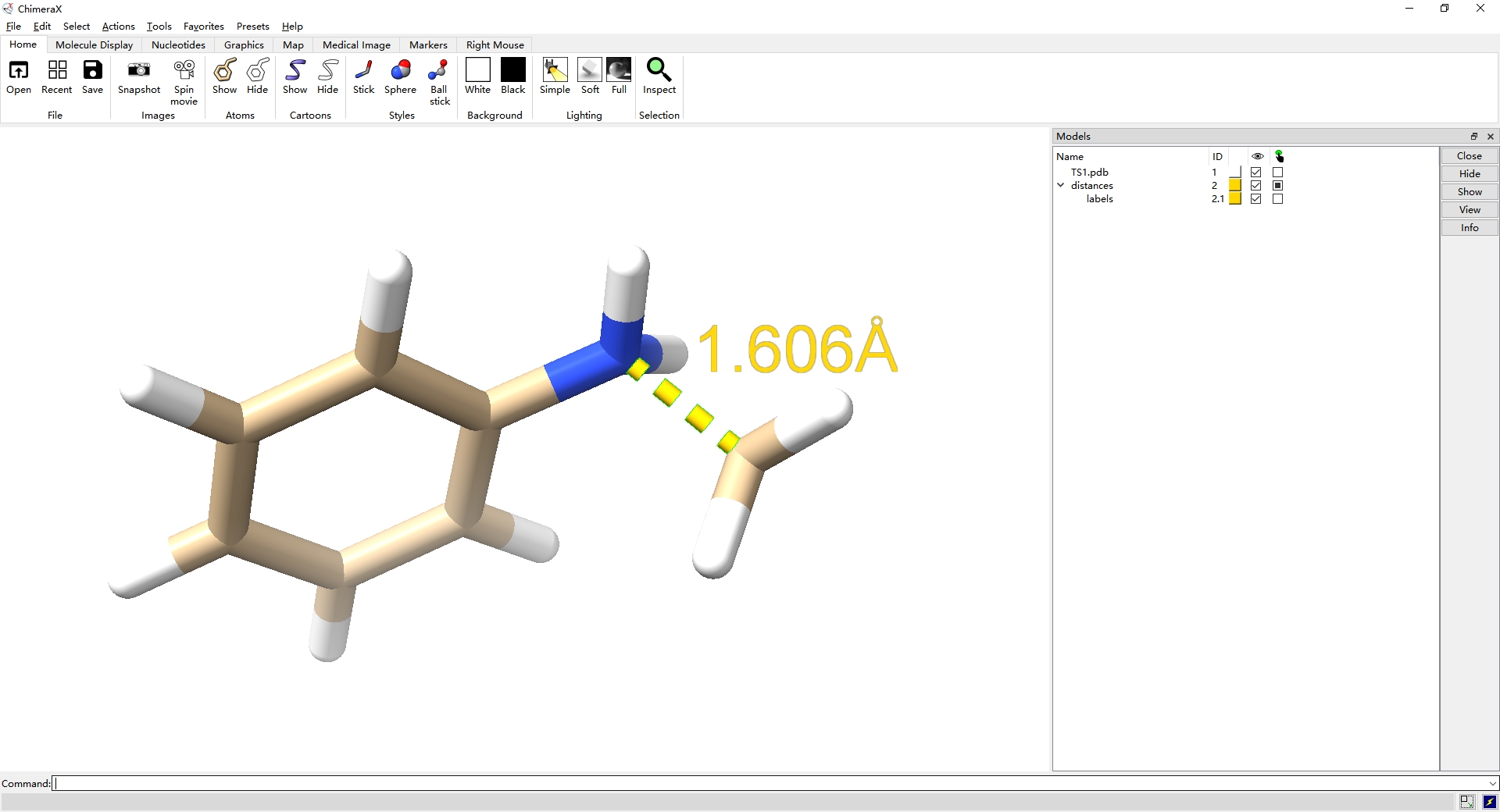
第八步,点击File下的Save…保存文件,建议保存一份文件名为cxs的ChimeraX文件,再保存一份文件名为bmp或者png的图片文件。(这里保存为bmp文件)
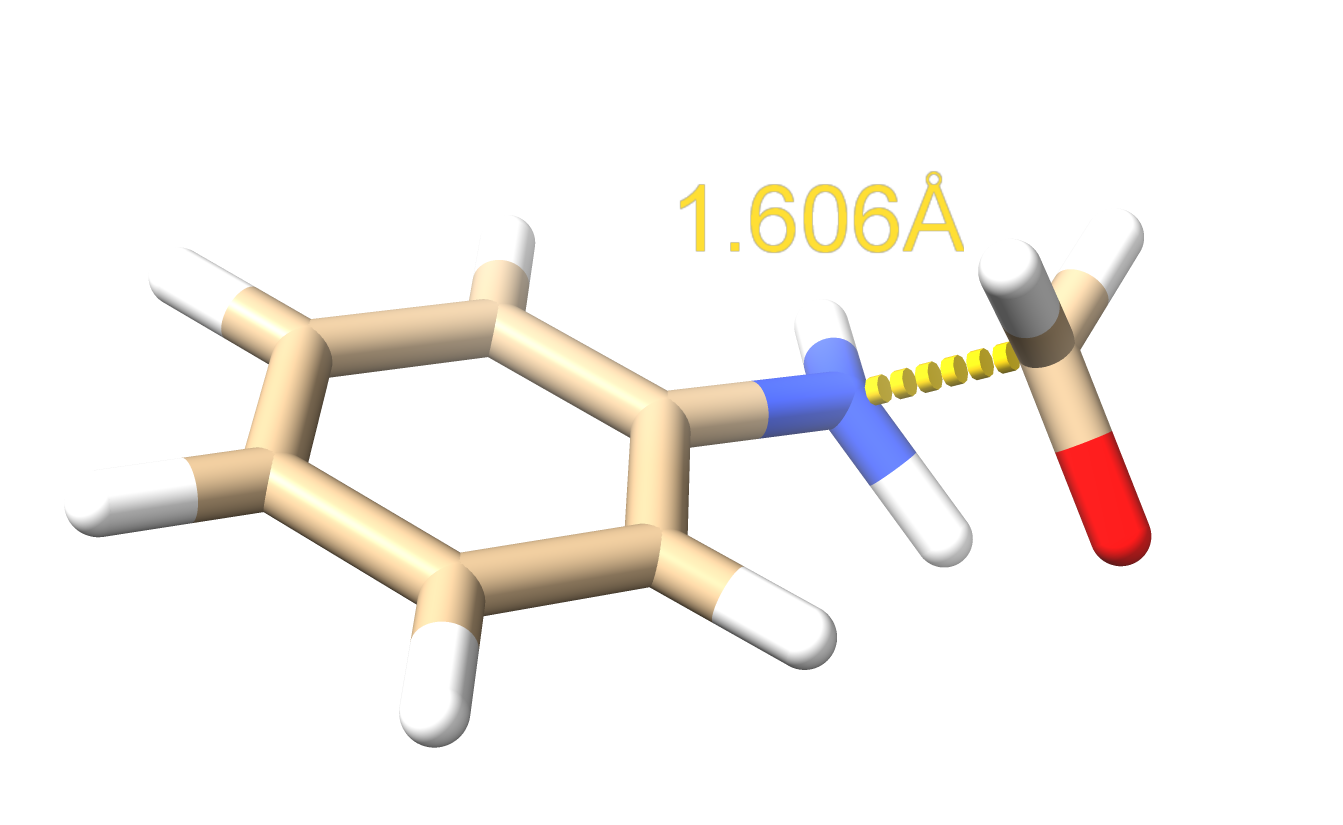
以上操作就算完成了最开始的的想法,画出一个过渡态的伪键,并标记其键长。对于其他的分子同理操作。
修改后的反应机理图
最后
ChimeraX作为一款功能强大的生化分析和可视化软件,对于我现在使用的是有点大材小用了,但现阶段也仅仅是使用它重画了分子结构(用C4D太麻烦了,也不太会,而ChimeraX的分子渲染风格是美观的)。以上记录笔记仅仅是简单的文件导入和图片的导出操作,关于软件的其它操作,等学会了再补充进来。