最近看了Tim(B站UP主)的一些视频后,产生了写一些内容的想法,以实现以写促学的目的。现阶段,我在计算方面的学习止步不前,还停留在吃老本的阶段(虽然没有什么老本可吃),但又想再学习一些新东西的想法,因此,开始想写一些计算的知识点整理(仅为DFT计算),一是帮助有可能看到这篇博文的读者,二是巩固自己的对这些内容的掌握。所写内容可能不成体系,没有前后逻辑关系,所以选择自己想看的部分吧。这部分主要写结构优化的部分,涉及分子结构的构建、任务的提交和结果的查看。
什么是结构优化
结构优化是指对整个输入体系的坐标进行调整,得到一个相对稳定的基态结构。我们知道基态是一个稳定态,但是对于一个基态势能面,我们需要去找一个最稳定的点,也就是所谓的极小值点(你可以想象在一个凹凸不平的平面上找那里面的凹陷点)。但是对于整个势能面来说,势能面上不止一个极小值点,因此有必要去找最小值点(这个点的能量最低,也意味着更稳定,但这不是一个必要条件,需要根据情况去选择最优结构)。
在Gaussian中结构优化的关键词为opt。
而结构优化根据优化需求分为非限制性优化和限制性优化,常规优化一般为非限制性优化,即基于所有原子的优化。
分子结构的构建
手搓版
对于没有晶体结构或者没有报道结构信息的分子,这种分子结构就需要通过建模软件手搓,常用的手搓软件有GaussView和Avogadro(推荐前者)。
关于如何使用GaussView可以见《基于Gaussian理论计算的粗浅经验小结》中的GaussView软件的操作。
非手搓版
对于那些有晶体结构的分子,可以通过获取晶体结构的xyz坐标来作为优化的分子坐标。而对于那些已被报道的结构信息分子,可以通过文献获取相应的分子坐标。
当然也可以通过对这些结构进行修饰来获取目标结构,这将极大的减少工作量,尤其是具有立体空间关系的分子,如配合物、分子笼、MOFs。
任务的提交
在提交任务之前,要对分子结构进行任务的设置。
对于非限制性结构优化的分子,只需要使用结构优化的关键词opt即可。而对于限制性结构优化的分子,则需要考虑原子冻结的问题。对于原子冻结来说,一般分为两种方法。
方法一:
对想冻结的原子可以在文本文件的坐标部分进行修改,如想冻结原子1,在原子1符号和原子1坐标之间添加-1即可,对于不冻结原子,添加数字0即可。形式如下。
1 | %chk=molecular1.chk |
当然如上的冻结方式也可以通过GaussView进行操作。
首先将输入文件导入GaussView,使用原子选择工具选择需要冻结的原子。接下来点击分子窗口的原子,选中后该原子为亮黄色。这时点击Tools下的Atom groups…选项,显示如下窗口界面。这时需要将Atom Group Class选择改为Freeze,以及选中Freeze (Yes),点击Group Actions … 中的Add Selected Atoms to “Freeze (Yes)”。
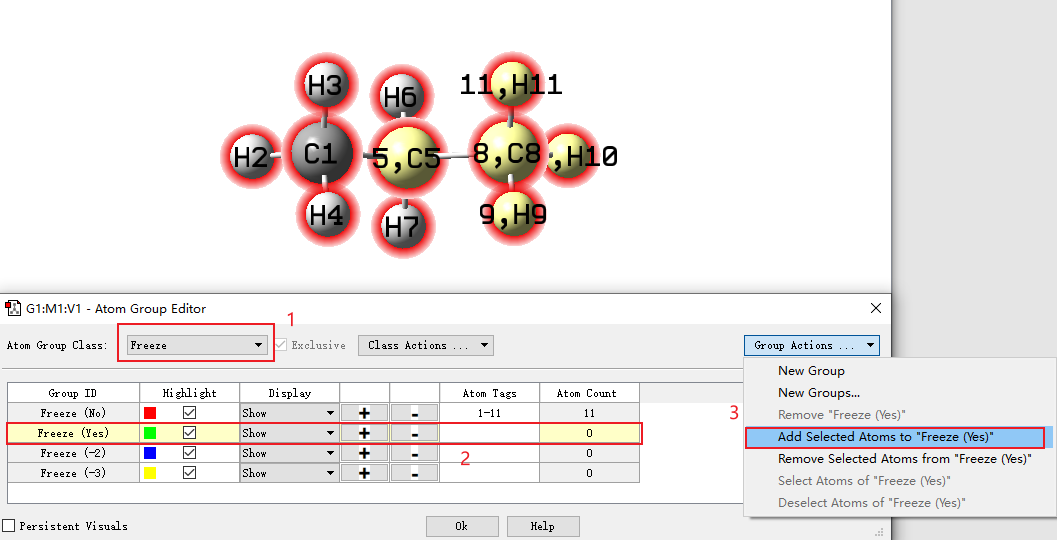
冻结完原子后,可以看到如下图中的Freeze (Yes)行中的Atom Tags中发现已经有了对应的原子序号。这时关闭Atom Group Editor窗口,保存文件即可。(对于冻结原子显示绿色的外发光,而不冻结原子显示红色外发光)
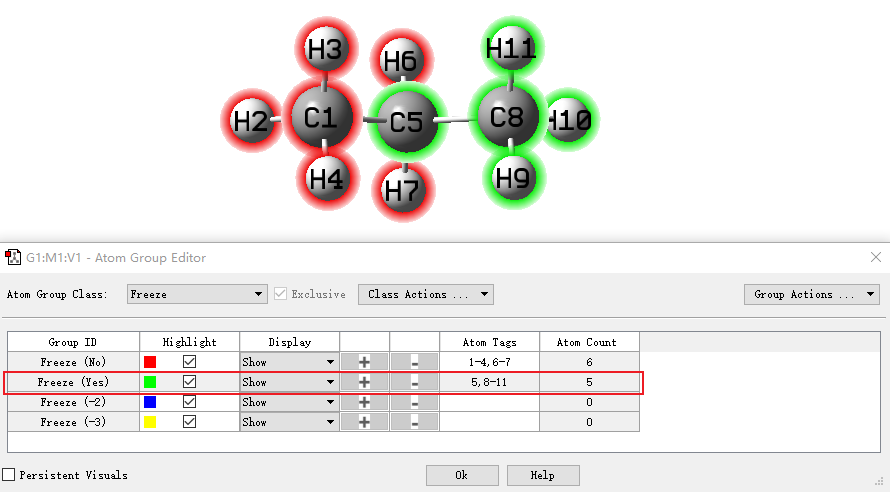
查阅输入文件的文本形式,也可以发现在原子符号和原子坐标之间添加了0和-1,说明已经冻结了想要冻结的原子。
方法二:
对于一类原子时,就可以使用一个讨巧的方法,在关键词opt后添加readopt选项,这时只需要在原子坐标后空一行然后用notatoms=冻结原子元素符号1,冻结原子元素符号2,冻结原子元素符号3来冻结原子即可。形式如下。
1 | %chk=molecular1.chk |
对于上面两种方法,建议根据实际情况来选择,如果是冻结部分基团,那么方法一更加灵活,如果是冻结好几种元素,只优化一种元素时,方法二则更便捷。
完成上面操作后,就可以像其他任务一样,将任务提交到对应的服务器,当然自己最好对输入文件设置内存和核数的多少,这样有利于对服务器资源的管理于分配。如果不会任务的提交,请查阅《基于Gaussian理论计算的粗浅经验小结》。
注:一般在进行优化计算的时候,都会写上freq,同时进行频率计算,以方便验证计算结果得到的结构是否为最优结构,如果没有虚频说明这个可用。但是否符合自己的预期,就需要根据自己的需要来判断是否可以被自己用于接下来的计算。
结果的查看
计算任务结束后,就可以得到一个out文件或者log文件(这两个都是日志文件,记录计算输出的信息)。
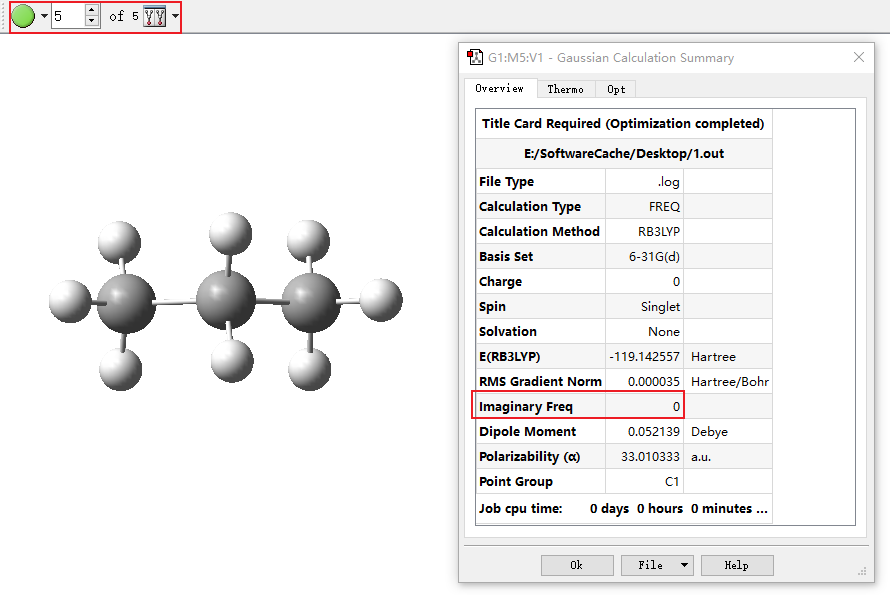
将一个计算完的out文件拖入GaussView后,就可以看到优化后的结构,以及优化了多少步,同时在分子结构窗口点击鼠标右键点击Results中的Summary…,就可以看到优化完后的一些分子信息,包括计算的方法和基组、分子的自旋多重度和能量,以及在这里最关心的虚频数。
如果是0,则说明这个结构可用,否者就需要重新的调整分子进行结构优化。
最后
本文介绍的内容是所有计算最开始的工作,如果不能获得一个优化好的结构,那么对于后续的所有计算都存在不合理性,对于数据的可信度也就有待商榷了。本文整理的内容比较简单,但对于大多数结构优化的任务,就是这样的操作,希望对能够读到这篇博文的人有所帮助。